
Your Historical Experiment Data Is Worth More Than You Think — Here's How AI Unlocks It
Most chemical R&D teams have a problem they don't talk about.
Somewhere in your organization — in a shared drive, a lab notebook, an ERP system, or a folder of Excel files nobody has opened in two years — there are hundreds, sometimes thousands, of experiment records. Reaction conditions. Batch results. Formulation attempts. Performance measurements.
You ran those experiments. You paid for them. You documented them.
And then you moved on to the next problem, leaving all of that data behind.
Here's what that data actually is: a predictive model waiting to be built. Every data point you generated in the lab is a piece of evidence about how your chemical system behaves. Together, they contain patterns — correlations between input conditions and output performance — that no human analyst would ever catch by scrolling through a spreadsheet.
ChemCopilot exists to extract those patterns. Bring your historical data. We build the AI models that turn it into decisions.
The Hidden Cost of Unused R&D Data
A pharmaceutical formulation team once came to us with a challenge: they needed to improve yield consistency in a batch crystallization process. They estimated it would take three to four months of new experiments to gather enough data to make a confident decision.
When we asked about historical records, they found 340 rows of past batch data going back two years. They had never looked at it analytically.
Within 48 hours of ingesting that data into ChemCopilot, the machine learning model identified that crystallization temperature and power input — not the variables the team had been focusing on — were the dominant drivers of particle size consistency. The team hadn't seen this because no single person had been able to hold all 340 rows in their head at once.
The four-month experiment plan became a two-week targeted validation. The historical data had the answer the whole time.
This is not an unusual story. It is the most common story in chemical R&D.
Step 1: Bring Your Data — In Whatever Format It Lives In
The first question most R&D teams ask is: "Do we need to clean and structure our data before we can use it?"
The short answer is: not as much as you think.
ChemCopilot is designed to ingest data from the formats your team already uses — not an idealized version of what your data should look like.
What you can bring:
Excel and CSV files — batch records, formulation logs, reaction screening results, quality control measurements. If it's in a spreadsheet, ChemCopilot reads it.
Research papers and patents in PDF — internal technical reports, published literature relevant to your chemistry, competitor patents. Our AI/OCR layer extracts structured information from text, tables, and figures.
Handwritten lab notes — digitized through our document processing pipeline. Even legacy records from notebooks count.
API connections from MES/ERP systems — if your manufacturing execution system or enterprise resource planning platform holds production data, ChemCopilot connects directly without requiring a manual export.
The goal is to remove the friction between where your data lives and where it needs to go. Because the more data you bring, the better the models get — and any barrier to ingestion is a barrier to insight.
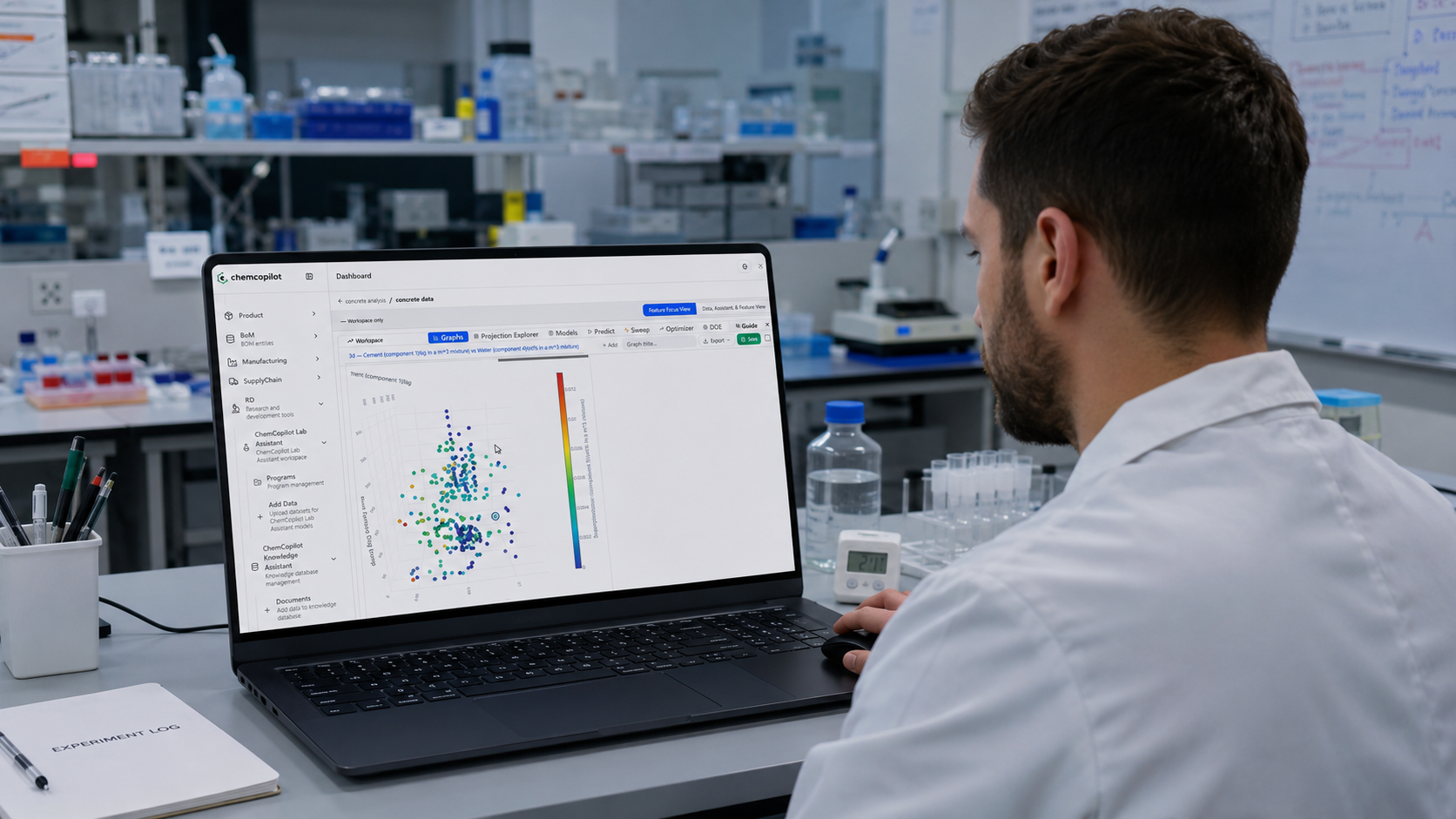
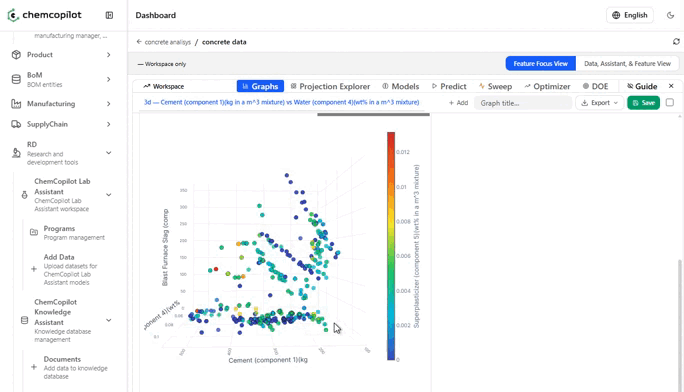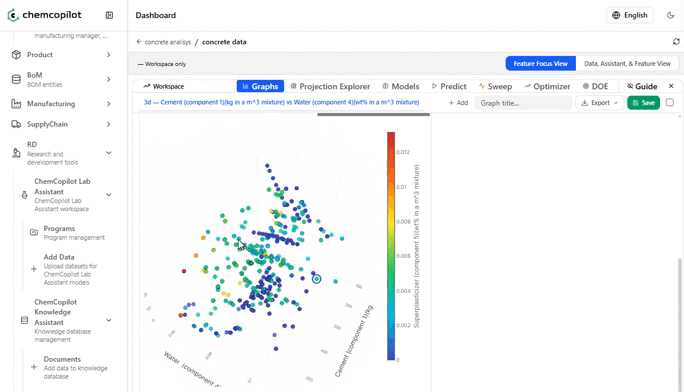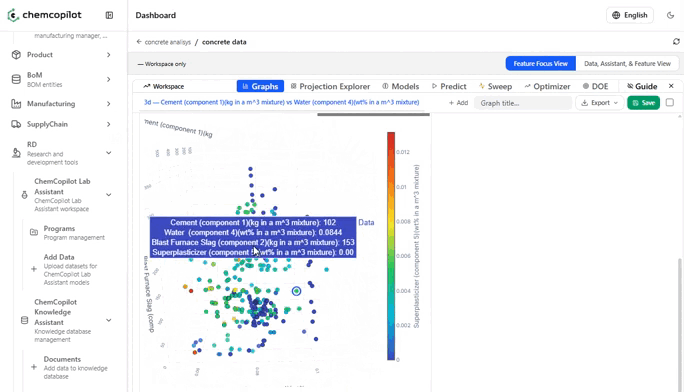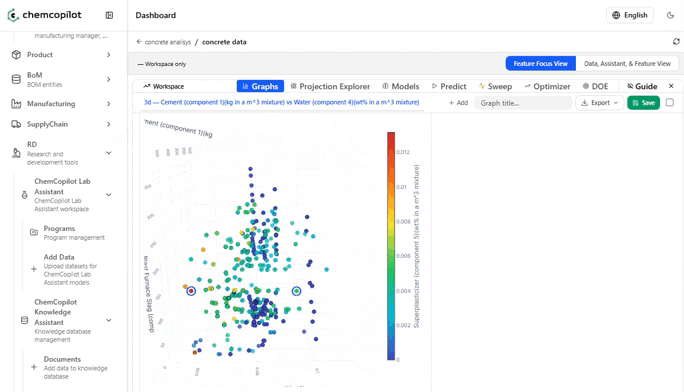
Step 2: Visualize, Then Model
Once your data is in, the first thing ChemCopilot does is show it to you.
This sounds trivial. It is not.
Most R&D teams have never seen their historical data displayed graphically as a complete dataset. Scatter plots of input vs. output variables. Correlation heatmaps across every measured parameter. Histograms of how your conditions were distributed across experiments. These visualizations alone — before any model is built — routinely surface insights that had been invisible for years.
You see which variables you've been over-testing. Which ranges you've never explored. Which combinations of conditions tend to cluster around your best results.
Then comes the modeling.
ChemCopilot's machine learning suite lets you train predictive models on your data without writing a single line of code. You select your input variables — temperature, reagent concentrations, reaction time, whatever your process involves — and your output target — yield, purity, particle size, viscosity, cost — and the platform evaluates multiple model architectures to find the best fit for your specific dataset.
Available architectures include:
XGBoost — the industry standard for tabular chemical data. Fast, accurate, and excellent at capturing nonlinear relationships between process variables.
Random Forest — robust for smaller datasets and particularly useful when you need to understand which variables matter most, not just get a prediction.
Neural networks — for larger, more complex datasets where the relationships between inputs and outputs are highly nonlinear.
Gradient Boosting and Elastic Net — for specific use cases where interpretability or regularization matter.
The platform automatically evaluates model quality using R² and cross-validation, so you know whether the model is ready to make reliable predictions — or whether you need more data in certain regions of your experimental space.
The most valuable output at this stage is often not the model itself: it is the feature influence analysis. A ranked list of which input variables are most predictive of your output. This tells your team where to focus their next experiments — and, equally important, where to stop running experiments that won't move the needle.
Step 3: Run AI Agents to Simulate, Optimize, and Design
With a trained model in place, ChemCopilot's AI agents take over the heavy computational work so your scientists can focus on decisions, not calculations.
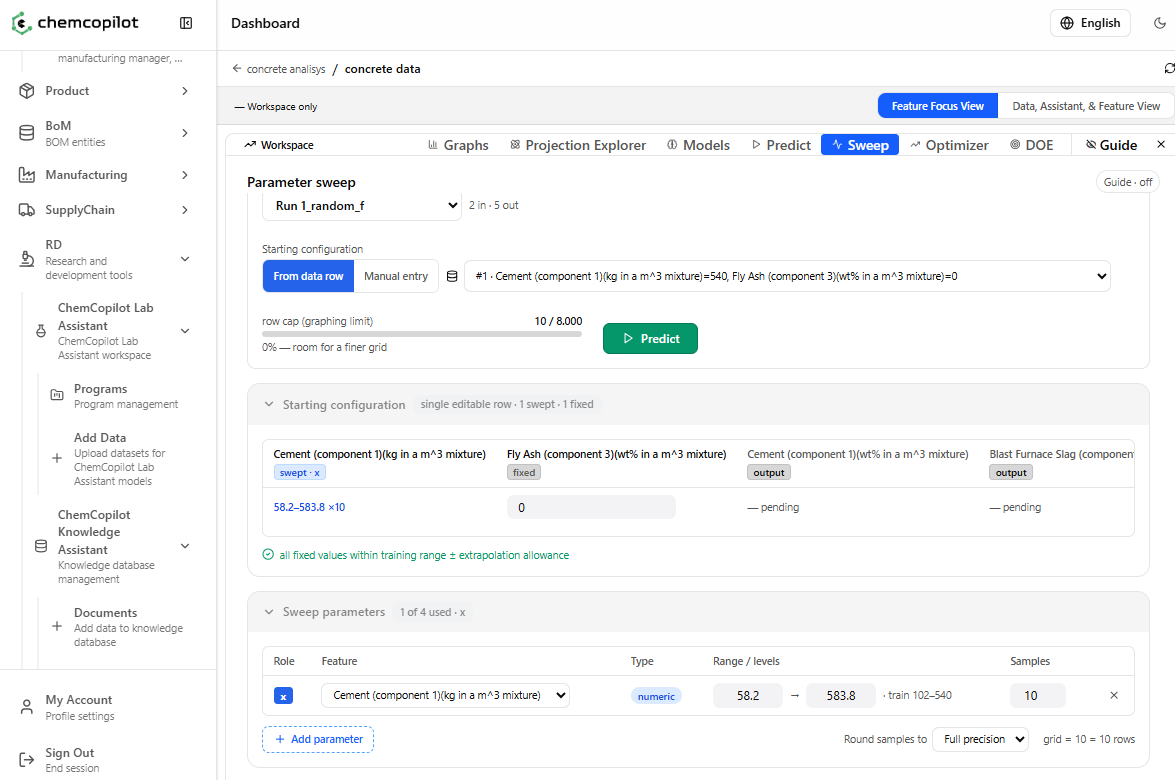
In-silico simulations let you run thousands of virtual experiments in seconds. Instead of testing twenty temperature conditions in the lab, you ask the model: what does my output look like across this entire temperature range? The answer arrives in a parameter sweep — a complete map of predicted performance across any combination of input variables you define.
Inverse design and optimization work the opposite direction. You specify the output you want — a target yield of 92%, a viscosity between 800 and 1200 cP, a minimum purity of 99.5% — and the optimization algorithm searches your input space to find the conditions most likely to hit that target. It is not a guarantee. It is a ranked list of experiments worth trying first, ordered by predicted probability of success.
Design of Experiments (DOE) generation helps teams that are starting a new project with minimal data. ChemCopilot uses your defined variable ranges to design a statistically efficient set of initial experiments — reducing the number of runs needed to build a good model from scratch by an order of magnitude.
Molecular-level simulations extend the platform into the structure-property domain — using SMILES representations and chemical embedding strategies to model how molecular structure affects physical and functional properties. This is particularly powerful for formulation scientists and medicinal chemists who need to screen candidate molecules before committing to synthesis.
What This Means for Your R&D Timeline
The compounding effect of these three steps — ingest, model, simulate — is not incremental improvement. It is a categorical change in how fast your R&D team can move
The experiments you would have run to find the answer have already been run. They are sitting in your data. ChemCopilot finds the signal in them and tells you where to look next.
Teams using this approach consistently report:
Reduction in required experimental runs of 60–90% for optimization problems where sufficient historical data exists
Identification of dominant process variables within hours of first model training, rather than months of targeted experimentation
Faster regulatory documentation because every model input, prediction, and decision is logged with a complete audit trail
The time you save is not just time. It is cost. It is competitive advantage. It is products reaching market faster and experiments that would have failed getting caught in silico instead of in the lab.
Getting Started
You do not need a clean, perfectly structured dataset to begin. You need what you already have.
Upload your historical batch records. Connect your ERP. Share your technical reports. ChemCopilot handles the ingestion, builds the models, and puts the insights in front of your team — in minutes, not months.
The data you generated over the last two, five, or ten years has been waiting for this.
ChemCopilot is an AI-native R&D platform for chemical formulation, process development, and regulatory compliance. Built for R&D teams in specialty chemicals, pharmaceuticals, coatings, polymers, and beyond.