
Drug Design Principles and Toxicity Prediction in the Age of AI - 2026+
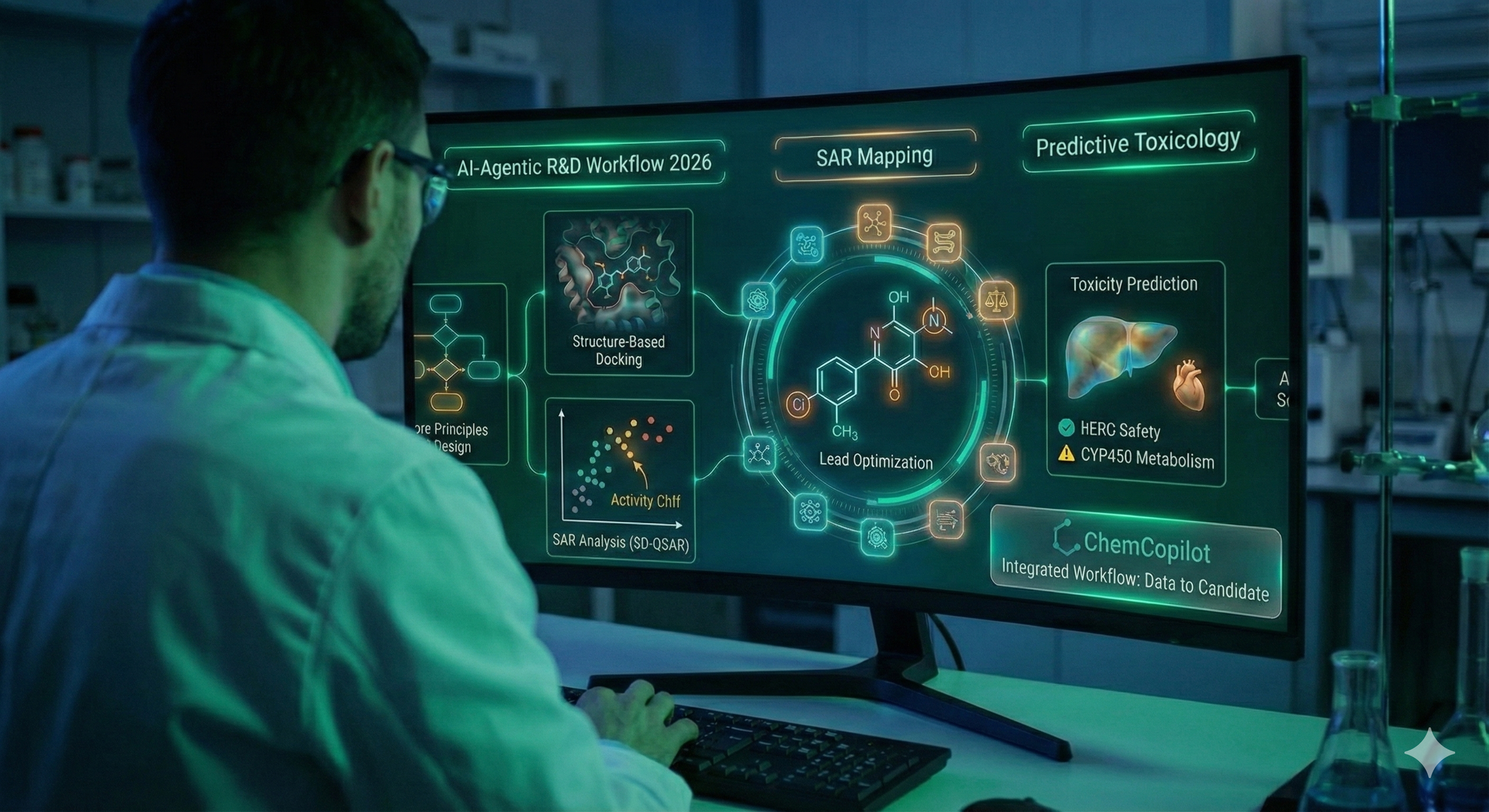
The evolution of drug discovery has shifted from serendipitous discovery to a highly structured, data-driven discipline. In 2026, the integration of Lead Optimization, Structure-Activity Relationship (SAR) analysis, and AI-driven screening has reduced the time to clinical trials by nearly 40% compared to a decade ago.
1. Core Drug Design Principles: The Optimization Journey
Drug design begins with a "hit"—a molecule showing basic activity against a target. The goal of Lead Optimization is to transform that hit into a drug candidate that is not only potent but safe and deliverable.
Rational Design & Bioisosterism: Modern optimization heavily utilizes bioisosteres—atoms or groups with similar physical or chemical properties that produce broadly similar biological effects. Swapping a carboxylic acid for a tetrazole, for example, can improve bioavailability while maintaining target affinity.
ADMET-First Approach: Historically, toxicity was tested late. In 2026, ADMET (Absorption, Distribution, Metabolism, Excretion, and Toxicity) profiles are modeled simultaneously with potency.
Structure-Based vs. Ligand-Based: While X-ray crystallography remains a staple, Cryo-EM and AlphaFold-3 protein structures now allow for high-resolution docking in previously "undruggable" targets like intrinsically disordered proteins (IDPs).
2. SAR and QSAR: Mapping the Chemical Landscape
Structure-Activity Relationship (SAR) is the cornerstone of medicinal chemistry. It involves systematically modifying a molecule to see how its "shape" dictates its "function."
The Activity Cliff: SAR identifies "cliffs" where a minor change (e.g., adding a methyl group) leads to a massive jump in potency or a total loss of activity.
Quantitative SAR (QSAR): This uses mathematical models to relate chemical structure to biological activity. In 2026, QSAR has evolved into 3D-QSAR, which accounts for the spatial orientation of functional groups, providing a more "human-like" understanding of molecular fit.
Scaffold Hopping: When a lead compound has a problematic core (e.g., high toxicity or patent encumbrance), SAR data allows chemists to "hop" to a different core scaffold that maintains the same spatial arrangement of key functional groups (pharmacophores).
3. AI-Powered Virtual Screening and Toxicity Prediction
The most significant leap in Active Pharmaceutical Ingredient (API) development is the transition from physical screening to AI-native virtual screening.
Deep Learning and Graph Neural Networks (GNNs)
Traditional docking was computationally expensive. Modern Graph Neural Networks treat molecules as mathematical graphs (atoms as nodes, bonds as edges), allowing AI to "screen" libraries of billions of compounds in hours. These models don't just find "fits"; they predict binding free energy with chemical-grade accuracy.
Predictive Toxicity & "Safety-by-Design"
AI now predicts toxicological endpoints long before a molecule is synthesized:
hERG Inhibition: Predicting cardiotoxicity by modeling how a molecule interacts with the heart’s potassium channels.
CYP450 Metabolism: AI predicts which liver enzymes will break down the API, preventing dangerous drug-drug interactions.
Genotoxicity: Deep learning models identify "structural alerts" that suggest a molecule might damage DNA.
4. The 2026 R&D Ecosystem: ChemCopilot and Agentic Workflows
In the current landscape, tools like ChemCopilot have moved beyond simple chatbots. They act as Agentic AI, orchestrating complex R&D workflows:
Automated Literature Synthesis: Analyzing thousands of patents to ensure a new SAR direction is "white space."
Predictive Retrosynthesis: AI doesn't just design the API; it tells the chemist exactly how to build it using the most sustainable (Green Chemistry) routes.
Real-time Collaboration: Integration with Private Language Models (PLMs) ensures that sensitive molecular data remains secure while providing researchers with real-time feedback on lead viability.
Key Takeaways for 2026
Lead Optimization is no longer a linear path but a multi-objective optimization (MOO) problem solved by AI.
SAR Analysis has been augmented by 3D-QSAR and high-fidelity simulations.
AI for APIs has shifted the bottleneck from "finding a hit" to "optimizing for safety," significantly reducing the "attrition rate" in clinical trials.