
The Silicon Lab: How AI Models are Decoding Tabular Chemistry

In the modern laboratory, the most powerful instrument is often not a spectrometer or a centrifuge, but a computer. The transition from physical trial-and-error to predictive modeling has accelerated drug discovery, materials science, and toxicology.
At the heart of this revolution are specific machine learning architectures tailored for tabular chemistry data—datasets where molecules are represented by numerical "descriptors" (like molecular weight, solubility, or electronegativity).
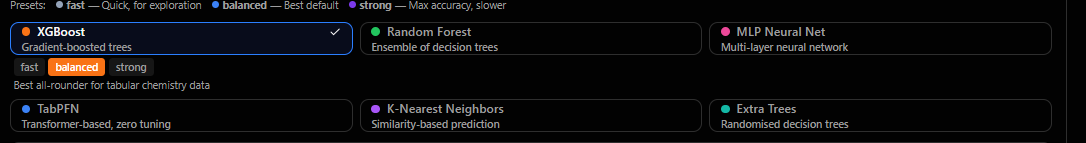
New Chemcopilot Features
1. The Heavyweight Champion: XGBoost
Category: Gradient-Boosted Trees
Role in Chemistry: The "Best All-Rounder"
XGBoost (Extreme Gradient Boosting) has become the industry standard for tabular data. In chemistry, it excels at predicting properties like melting points or binding affinity.
Why it works: It builds an ensemble of decision trees sequentially, where each new tree corrects the errors of the previous ones.
Chemistry Edge: It handles "missing data" gracefully—a common issue in experimental datasets where not every molecule has been tested for every property.
2. The Reliable Ensembles: Random Forest & Extra Trees
Category: Bagging-based Decision Trees
Role in Chemistry: Stability and Feature Importance
While XGBoost is fast and precise, Random Forest and Extra Trees are the workhorses of chemical informatics.
Random Forest: Creates a "forest" of independent trees and averages their results. It is incredibly difficult to "overfit," making it safe for small chemical datasets.
Extra Trees (Extremely Randomized Trees): Takes randomness a step further by choosing split points at random. This makes it faster and often better at handling the "noise" inherent in physical lab measurements.
3. The New Frontier: TabPFN
Category: Transformer-based
Role in Chemistry: Zero-Tuning Predictions
TabPFN represents a paradigm shift. Traditional models need to be "trained" on your specific data, which takes time. TabPFN is a Prior-Data Fitted Network—a transformer model that has already "learned" how to learn from tabular data.
The Advantage: It provides high-accuracy predictions instantly without hyperparameter tuning. For a chemist with a small dataset of 100 novel compounds, TabPFN can provide "Strong" model performance with "Fast" model speed.
4. Deep Learning: MLP Neural Nets
Category: Multi-Layer Perceptrons
Role in Chemistry: Non-Linear Complexity
Multi-Layer Perceptrons (MLPs) are the classic "AI" architecture. In chemistry, they are used when the relationship between structure and function is highly non-linear and complex.
Application: Often used in QSAR (Quantitative Structure-Activity Relationship) modeling, where subtle changes in a molecule's shape lead to massive changes in biological activity.
5. The Logic of Similarity: K-Nearest Neighbors (KNN)
Category: Instance-based Learning
Role in Chemistry: Similarity-based Prediction
KNN is the most intuitive model for a chemist. It operates on a simple principle: "Similar molecules have similar properties."
How it works: It maps molecules into a multi-dimensional space. To predict the toxicity of a new compound, it looks at the $K$ closest molecules already in the database and averages their values.
| Preset | Goal | Typical Model | Use Case |
|---|---|---|---|
| Fast | Exploration | KNN / Extra Trees | Screening millions of virtual compounds for a "rough idea." |
| Balanced | Default | XGBoost | Standard property prediction with reliable accuracy. |
| Strong | Max Accuracy | TabPFN / Optimized MLP | Final validation before heading to the physical lab for synthesis. |
Conclusion: The Smarter, Greener Future of Chemistry
The "Silicon Lab" isn't about replacing chemists; it's about narrowing the search space. By using models like XGBoost for stability or TabPFN for rapid-fire screening, researchers can skip thousands of failed experiments and focus their resources on the molecules most likely to change the world.
Whether you are optimizing a pharmaceutical formulation or designing a new sustainable polymer, these tools are the bridge between raw data and groundbreaking discovery.
🚀 Upcoming News: The ChemCopilot Evolution
We have some exciting news for the community! ChemCopilot is preparing to launch a new AI model very soon.
Integrated Power: This upcoming launch will feature a unified environment that includes all the models discussed in this article—from XGBoost to TabPFN.
Accessible Innovation: The model will be available for free, ensuring that every researcher and lab has access to the cutting edge of chemical intelligence.
Stay with us as we continue to build the tools that empower the next generation of scientific breakthroughs.